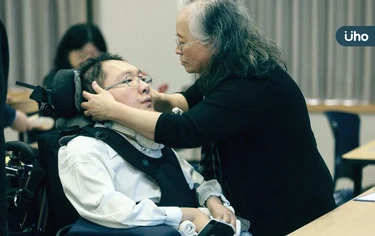
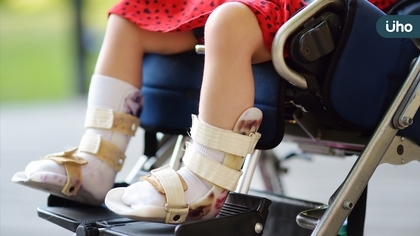
脊髓性肌肉萎縮症(SMA)是一種罕見的隱性遺傳性疾病,主要表現是神經運動元退化導致持續性肌肉退步、受傷,SMA發生率為一萬~四萬分之一,台灣約有400名案例。SMA臨床表現是全身肌肉退化,因此不僅是四肢與軀幹無力,最後連吞嚥、呼吸功能都受影響。與同樣為運動神經元退化的漸凍症比較,SMA在18歲之前發病者高達99%,因此對於家庭的照護衝擊也更大。 台北榮民總醫院兒童醫學部兒童神經癲癇科主任許庭榕醫師表示,SMA根據基因型態分為最嚴重的第一型、較為輕微的第二型、及最為輕微的第三型及第四型。以往尚未有藥物治療時,最嚴重的第一型為出生後六個月內發病,出生後往往除了四肢無力,更可能因為感染呼吸道疾病而無法維持生命,皆於兩歲前就會死亡;如果是出生後6到18個月發病的第二型或於於18個月後發病或青少年或成人期發病的第三型或第四型,也會因肌肉持續退化,可能導致無法行走,或影響走路,脊椎側彎或呼吸功能受影響等,影響日常生活甚巨。 SMA是少數透過提早治療能改變病程的罕病 許庭榕醫師提到,慶幸新藥發展,儘管SMA屬於罕病,但病患只要提早治療,不但有助延緩惡化,甚至有機會讓運動功能進步。目前的治療方式分為背針、基因治療、口服治療。其中,基因治療僅限6個月以下新生兒,占最大比例的18歲以下發病者以背針與口服藥物治療為主。而背針依治療指引在第一年需施打6劑,第二年以後治療穩定,每年只要僅需3次,回診治療同時預約下次施打時間,可大幅減少往返醫院次數與奔波之苦,便利性高。 許庭榕醫師說明,背針是目前臨床使用最成熟、累積最多實證研究的治療方式,安全性高,也相當有效。以往嬰兒確診為第一型SMA,幾乎無法存活至兩歲,但北榮就有案例是2018年出生的孩子,經由新生兒篩檢後確診的第一型SMA病童,透過背針治療的提早介入,到現在已經6歲,不僅病情穩定,甚至還可以跑跳,與一般健康孩子沒有兩樣;反觀同年齡未能提早就醫的孩子,因為病程已影響行走功能而必須輪椅代步,若能即時且提早治療,在神經未受傷嚴重時進快治療,之後的治療效果非常良好,可以與常人無異。 背針治療臨床運用成熟、效果佳 今年8月健保全面擴大背針與口服藥給付,許庭榕醫師提到,新藥已有相當好的進展及治療效果,對於成人病患而言,治療目標是避免運動功能繼續退步。他鼓勵病友都應積極治療,即便是成人或較晚接受治療病患,都可透過藥物延緩或預防神經肌肉持續退化,更有機會讓運動功能持續進步。 由於SMA病友需要相當長時間的復健與照護,因此跨科別的醫療團隊合作相當重要,以背針治療來說,即便是困難型脊椎側彎也能透過影像專科合作,不僅兼顧安全性,也讓治療效果發揮到最好。 許庭榕醫師強調,長期有效及穩定治療對SMA病友預後相當關鍵,SMA病友大部分在18歲之前就發病,都需要父母或專人照顧,除了耗費健保資源外,家長身體負擔及心理壓力都相當大,甚至可能必須辭掉工作照顧孩子,現在已經有治癒機會,不僅能夠減少經濟負擔,也讓病患的人生價值重回正軌。 許庭榕醫師也呼籲,SMA初始治療選擇也相當重要,雖然健保規範SMA病友終身僅能選擇一種藥物,但除非是對藥品有嚴重耐受不良或副作用,且經特殊專案審查核准,才有一次轉換機會。因此,鼓勵病友充分與醫師溝通治療計畫,選擇最適合自己的治療方式,及早接受治療且避免延誤最佳治療時機。